Research in the Corces Lab
In the Corces Lab, we aim to understand one fundamental question: why do some individuals develop neurodegenerative disease while others do not? The answer to this question, broadly speaking, boils down to "nature and nurture", or genetics and environment, and we approach both of these angles simultaneously by studying the noncoding genome and the epigenetic control of disease-related cellular phenotypes. While much of our work in the lab focuses on the two most common neurodegenerative diseases, Alzheimer’s disease (AD) and Parkinson’s disease (PD), we collaborate with other labs to study disease contexts ranging from traumatic brain injury to glaucoma.
Main focus #1: Noncoding genetics in neurodegenerative disease
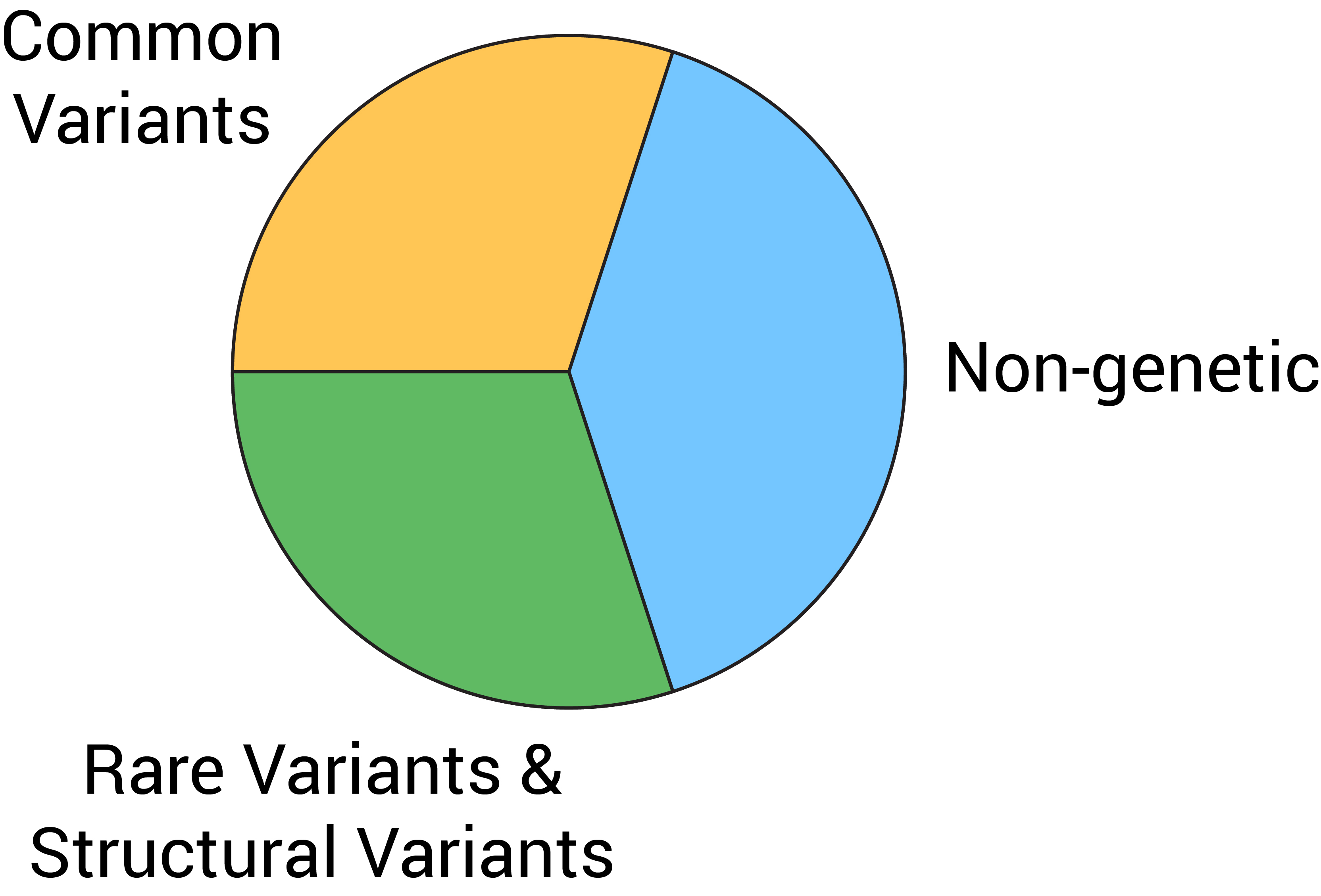
Despite decades of work correlating genetic variation to disease, the vast majority of disease predisposition remains unexplained. AD and other complex diseases are estimated to be 40-60% heritable. Yet our current best methods explain less than half of that predisposition. In practical terms, this means that for many patients, no therapeutics may be available or in the pipeline to counteract their disease. Why are we still missing so many of the genetic determinants of complex diseases? Though cliché, the streetlight analogy is highly applicable here – we tend to search for disease drivers among variants in the coding part of the genome, because it is easy to nominate the genes these variants disrupt. But coding regions represent approximately 2% of the human genome and the vast majority (> 95%) of variants map to noncoding regions of the genome!
The genetics of complex disease has largely been studied through the use of genome-wide association studies (GWASs). These studies have identified thousands of genetic variants, coding or noncoding, that correlate with an increased risk of developing AD or PD. However, few actionable therapeutic interventions have ensued. This is largely because GWASs are not specific enough: they home in on genomic loci associated with disease, but they do not pinpoint which specific variants are functional, nor do they identify the genes affected by variants that do not fall within a coding region.
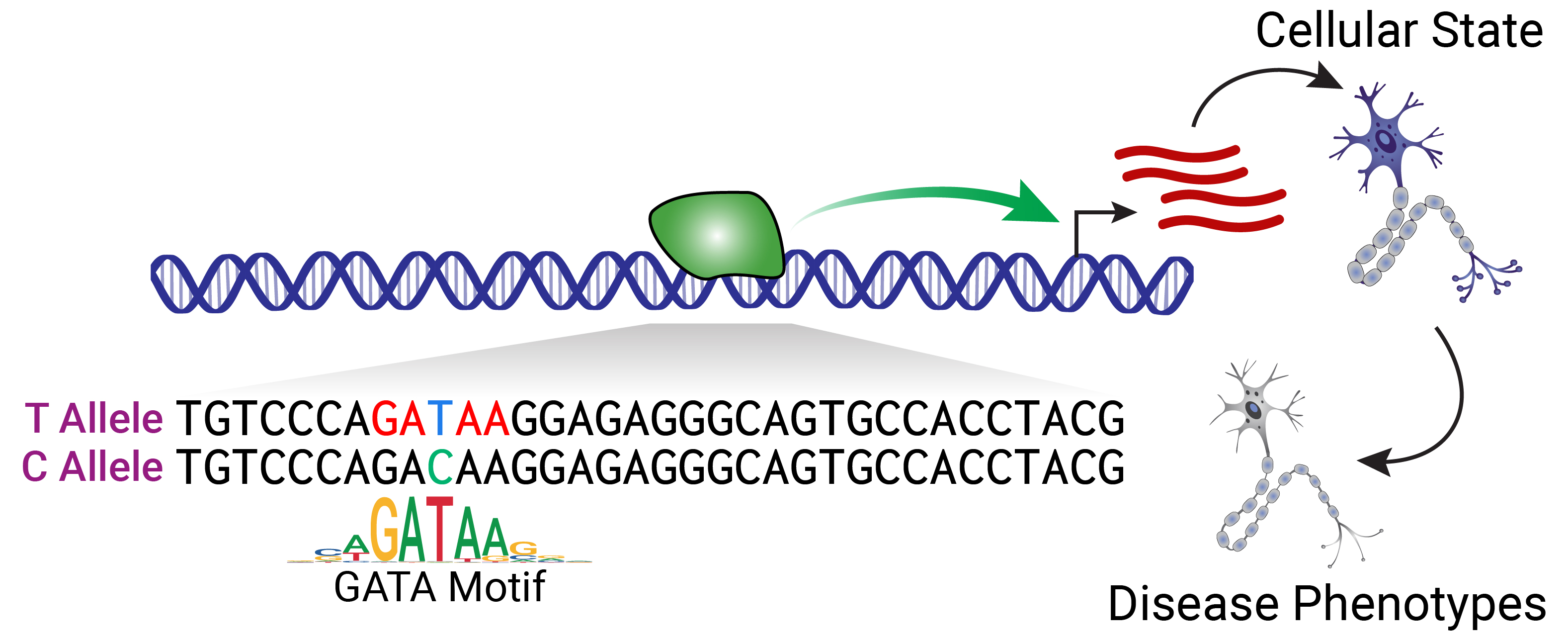
Noncoding variants primarily exert their effects by altering the sequence-specific binding of transcription factors (TFs), which ultimately changes where and when genes are expressed. A primary goal of the lab is to define the functional noncoding variants impacting predisposition to AD and PD. To this end, we integrate large-scale multi-omic profiling of patient tissues, state-of-the-art machine learning models, and high-throughput screens in iPSC-derived models of key brain cell types. By broadening our scope to the vast and underexplored noncoding genome, we expect to identify new genetic determinants of AD and PD pathogenesis and nominate new putative targets for therapeutic intervention.
Main focus #2: Epigenetics of disease
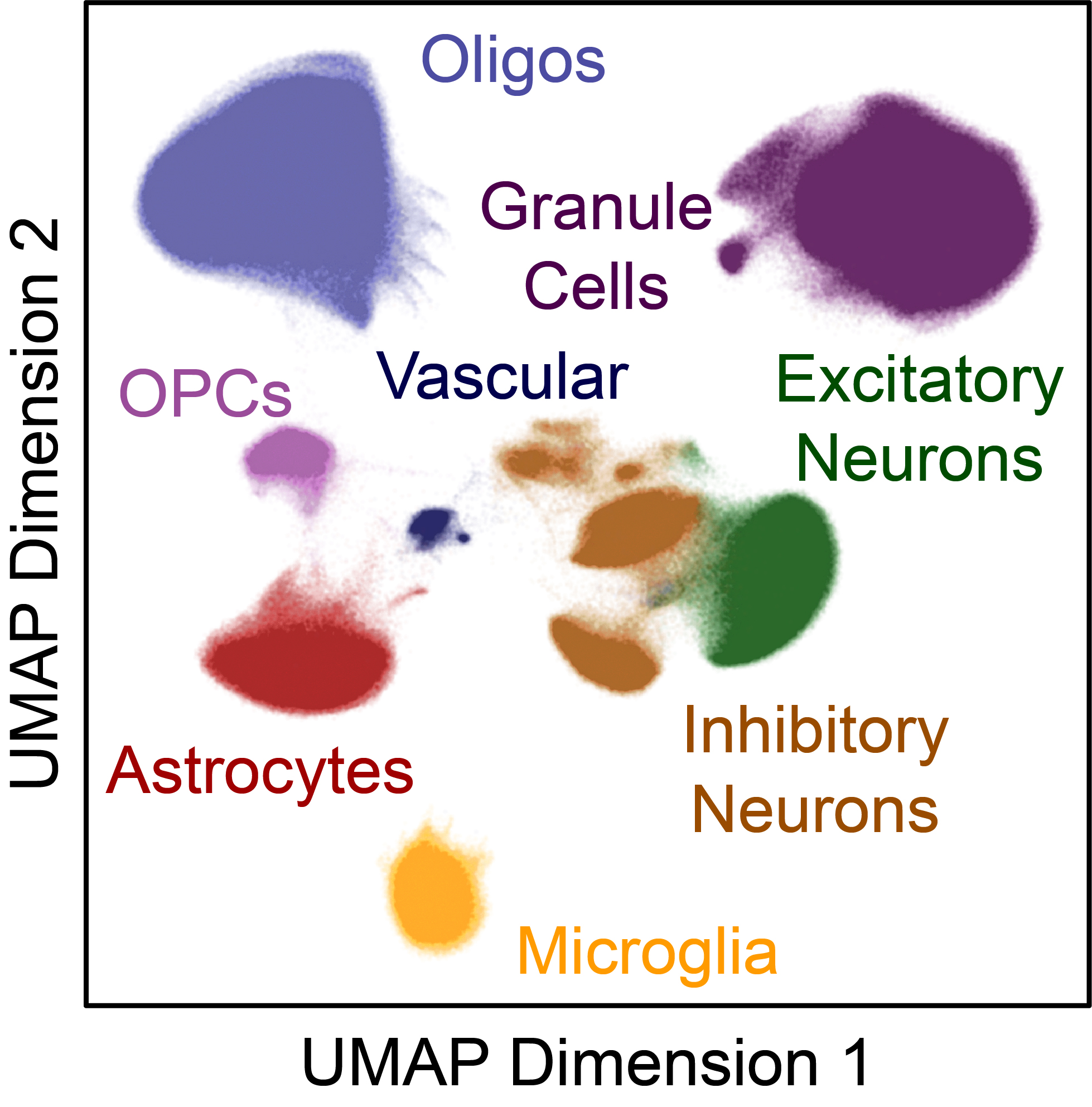
As discussed above, many AD and PD patients carry genetic variants that predispose them to disease since birth. Yet, most of them do not manifest symptoms until very late in life. This late onset indicates that non-genetic factors associated with aging or the environment contribute to disease processes. Such non-genetic factors impact our cells by changing how and when genes are expressed and they do this largely through altering the epigenome. However, it remains extremely challenging to disentangle how individual non-genetic factors relate to disease in human patients because such prospective studies are impractical. Instead, we hypothesize that environmental factors and aging (acting in concert with inherited genetics) converge on a small number of central disease mechanisms. To understand how these non-genetic factors contribute to disease, we profile the epigenomes and transcriptomes of postmortem patient tissue at single-cell resolution, searching for genes that are dysregulated in certain subsets of patients. Once we find features that appear to impinge on known disease mechanisms, we validate those using in vitro systems, leveraging large-scale CRISPR screens and molecular perturbations in iPSCs. Ultimately, we hope to one day relate those changes back to the original non-genetic factors that drove them to begin with.
Other areas of interest
Ancestry-specific genetic effects
Certain genetic variants exhibit different effects in different ancestral backgrounds. For instance, the APOE4 isoform, the strongest known common genetic determinant of AD, has a weaker effect on disease predisposition in individuals of African ancestry and a stronger effect in individuals of East Asian ancestry compared to individuals of European ancestry. This implies that other genetic loci modify an individual’s susceptibility to the effects of APOE4. In the lab, we are uncovering ancestry-specific modifiers of APOE4 and other disease-associated variants, and investigating whether they point to novel aspects of disease biology.
Rare variants
Rare variants have fueled therapeutic discovery across a wide array of diseases. In the case of AD, rare variants found in familial pedigrees have led to the identification of central disease drivers such as amyloid precursor protein or the presenilins. However, nearly all of the research to date has focused on rare coding variants, ignoring the more than 95% of rare variants that reside in the noncoding genome. This is because it has been challenging to predict the impact of noncoding variants and each individual harbors more than 10,000 rare noncoding variants, a daunting haystack in which to find the right needle. However, we now sit at a crossroad where technologies and datasets finally match the scale and complexity of the problem of rare noncoding variants. We hypothesize that rare noncoding variants drive disease particularly in the 40% of sporadic cases and nearly 50% of hereditary cases that do not harbor any of the known or putative AD-associated variants. Moreover, we believe that some rare variants may target categorically different genes than do common variants, because of the much larger effect sizes that can occur in rare variants. Using the same high-throughput methods that we have developed for prioritization of GWAS-defined disease-associated common variants, we are working to functionalize rare noncoding variants as potential drivers of disease in both AD and PD.
Resilience to AD pathology
AD is associated with the accumulation of beta-amyloid into plaques and hyperphosphorylated tau into neurofibrillary tangles. Though the precise mechanisms are still debated, these two pathologic changes are widely regarded as drivers of neuronal dysfunction and subsequent cognitive decline. However, some individuals appear resilient to the accumulation of AD-related pathology, maintaining unperturbed cognitive function for years. We are interested in understanding (i) if this resilient state truly exists or if these individuals would eventually develop AD-related cognitive impairment, and (ii) if resilience truly exists, how this resilience is genetically and epigenetically encoded. Ultimately, we hope to use our findings to increase resilience across more vulnerable patients.
Selective vulnerability of brain cells and regions to disease
In AD, only particular types of neurons in particular brain regions succumb to degeneration. Such selective vulnerability also characterizes other neurodegenerative diseases, even if the neuronal types affected differ. In most cases, the cells that accrue the most pathology are the most disrupted. However, many examples exist of neuronal cell types and brain regions that accumulate pathology but remain relatively healthy. We and others hypothesize that selective vulnerability (or resilience) to pathology results from both cell-autonomous properties of neurons and cell non-autonomous properties of surrounding glial cells that is reflected in their gene expression patterns. We are using single-cell transcriptomic approaches to characterize cell type- and brain region-specific differences that may underlie selective vulnerability.