Publications
For a complete list of publications see Google Scholar or PubMed.

Nature Protocols 2022
Grandi FC, Modi H, Kampman L, Corces MR*
The Assay for Transposase-accessible Chromatin using sequencing (ATAC-seq) has become a mainstay in genomics reseach. In this protocol manuscript, we describe in detail how to perform all aspects of bulk ATAC-seq and provide considerations for how to best prepare your input cells/nuclei. We highlight considerations for when and when not to use ATAC-seq and provide recommendations for analytical best practices. As part of this publication, we have also released scripts to perform the iterative overlap peak merging procedure that we recommend for creating a robust bulk ATAC-seq peak set.
[pubmed] [pdf] [Nature Protocols]
Nature Genetics 2021
Granja JM*, Corces MR*, Pierce SE, Bagdatli ST, Choudhry H, Chang HY†, Greenleaf WJ†
The advent of large-scale single-cell chromatin accessibility profiling has accelerated our ability to map gene regulatory landscapes, but has outpaced the development of robust, scalable software to rapidly extract biological meaning from these data. Here we present a software suite for single-cell analysis of regulatory chromatin in R (ArchR; www.ArchRProject.com) that enables fast and comprehensive analysis of single-cell chromatin accessibility data. ArchR provides an intuitive, user-focused interface for complex single-cell analyses enabling the analysis of over 1.2 million single cells within 8 hours on a standard Unix laptop.
[pubmed] [pdf] [Nature Genetics]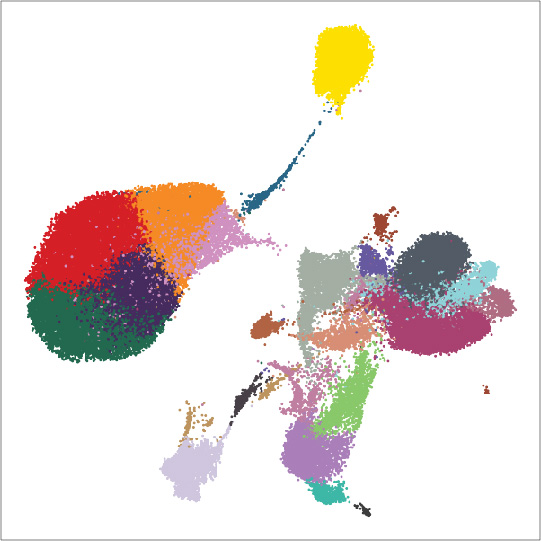
Nature Genetics 2020
Corces MR, Shcherbina A, Kundu S, Gloudemanns MJ, Fresard L, Granja JM, Louie BH, Eulalio T, Shams S, Bagdatli ST, Mumbach MR, Liu B, Montine KS, Greenleaf WJ, Kundaje A, Montgomery SB, Chang HY†, Montine TJ†
We develop a machine learning classifier to integrate multi-omic epigenetic data and predict dozens of functional SNPs, nominating gene and cellular targets for previously orphaned GWAS loci. These predictions both inform well-studied disease-relevant genes, such as BIN1 in microglia for Alzheimer’s disease (AD) and reveal novel gene-disease associations, such as STAB1 in microglia and MAL in oligodendrocytes for Parkinson’s disease (PD). This work greatly expands our understanding of inherited variation in AD and PD and provides a roadmap for the epigenomic dissection of noncoding regulatory variation in disease.
[pubmed] [pdf] [Nature Genetics]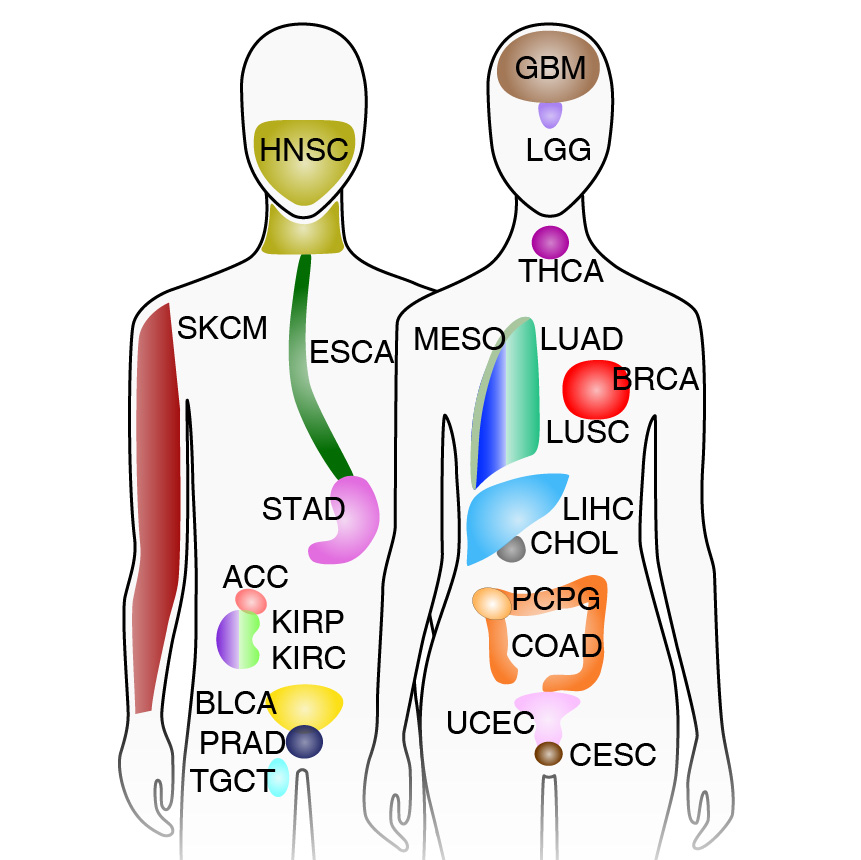
Science 2018
Corces MR*, Granja JM*, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, Satpathy AT, Mumbach MR, Hoadley KA, Robertson AG, Sheffield NC, Felau I, Castro MAA, Berman BP, Staudt LM, Zenklusen JC, Laird PW, Curtis C, The Cancer Genome Atlas Research Network, Greenleaf WJ†, Chang HY†
We present the genome-wide chromatin accessibility profiles of 410 tumor samples spanning 23 cancer types from The Cancer Genome Atlas (TCGA). We identify 562,709 transposase-accessible DNA elements that substantially extend the compendium of known cis-regulatory elements. Integration of ATAC-seq with TCGA multi-omic data identifies a large number of putative distal enhancers that distinguish molecular subtypes of cancers and nominates long-range gene-regulatory interactions in cancer. Moreover, these data pinpoint noncoding mutations that drive enhancer activation and may affect patient survival. These results suggest a systematic approach to understanding the noncoding genome in disease to advance diagnosis and therapy.
[pubmed] [pdf] [Science]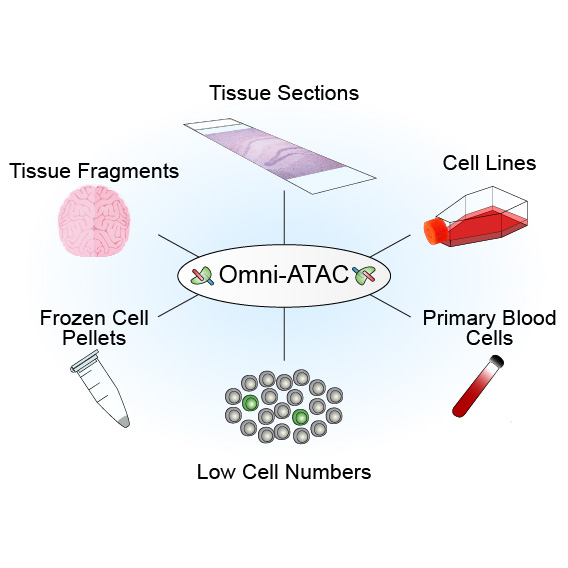
Nature Methods 2017
Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, Kathiria A, Cho SW, Mumbach MR, Carter AC, Kasowski M, Orloff LA, Risca VI, Kundaje A, Khavari PA, Montine TJ, Greenleaf WJ†, Chang HY†
We present Omni-ATAC, an improved ATAC-seq protocol for chromatin accessibility profiling that works across multiple applications with substantial improvement of signal-to-background ratio and information content. The Omni-ATAC protocol generates high-quality chromatin accessibility profiles from almost any starting material, from cell lines to archival frozen tissue samples and 50-µm sections. Moreover, Omni-ATAC saves money through fraction of reads in peaks and lower fraction of reads mapping to the mitochondrial chromosome.
[pubmed] [pdf] [Nature Methods]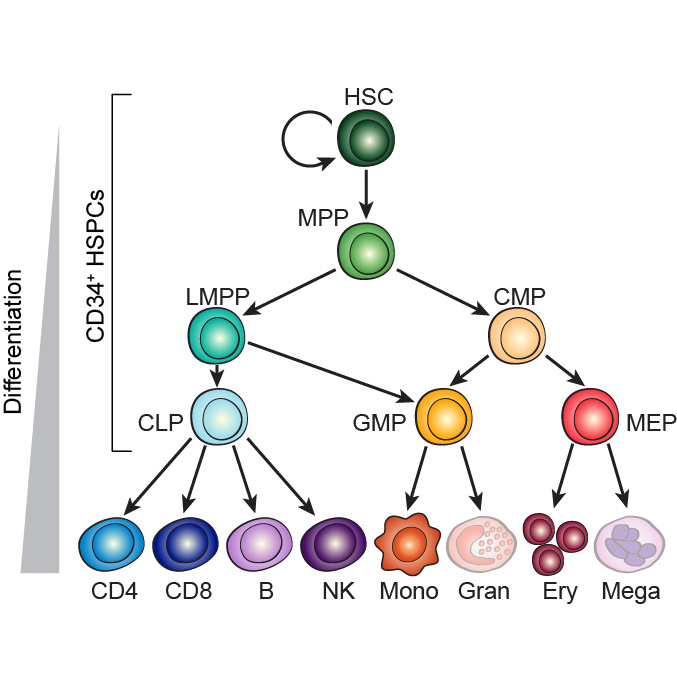
Nature Genetics 2016
Corces MR*, Buenrostro JD*, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, Majeti R†, Chang HY†
We define the chromatin accessibility and transcriptional landscapes in 13 human primary blood cell types that span the hematopoietic hierarchy. Exploiting the finding that the enhancer landscape better reflects cell identity than mRNA levels, we enable enhancer cytometry for enumeration of pure cell types from complex populations. We identify regulators governing hematopoietic differentiation and further show the lineage ontogeny of genetic elements linked to diverse human diseases. In acute myeloid leukemia (AML), chromatin accessibility uncovers unique regulatory evolution in cancer cells with a progressively increasing mutation burden. Single AML cells exhibit distinctive mixed regulome profiles corresponding to disparate developmental stages.
[pubmed] [pdf] [Nature Genetics]